
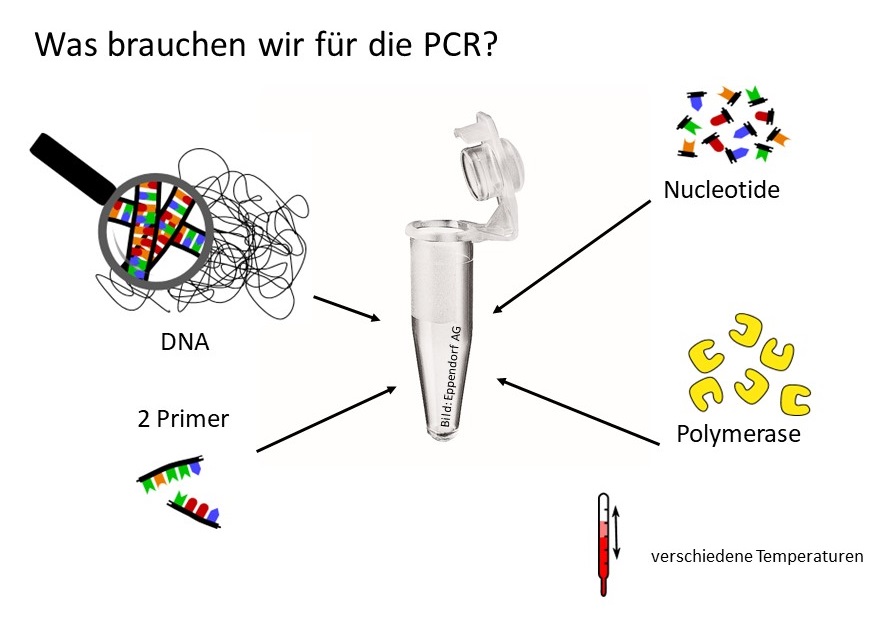
Die Polymerasekettenreaktion („Polymerase Chain Reaction“ = „PCR“) ist eine geniale Erfindung. Jüngere Wissenschaftler können sich gar nicht mehr vorstellen, wie Molekularbiologie ohne PCR überhaupt möglich war!
Das Konzept orientiert sich an der Natur: Bei jeder Zellteilung wird die DNA verdoppelt. Ein Prozess, der „Replikation“ genannt wird. Warum soll man das nicht auch im Reagenzglas machen können?


Die DNA-Polymerase, ein Enzym, das die Bausteine der DNA (Nukleotide) nach einer Vorlage zusammensetzen kann, spielt bei der PCR eine entscheidende Rolle. Diese DNA-Polymerase konnte man recht einfach in großen Mengen von E. coli-Bakterien synthetisieren lassen – ebenso wie heute das Insulin für Diabetiker in großen Mengen von Bakterien produziert wird.
Ein Problem der DNA-Polymerase ist, dass sie nur ein bereits vorhandenes Stück DNA verlängern kann, also ein Stückchen DNA als „Starter“ benötigt. Das Problem wurde gelöst, als man gelernt hatte, kurze Stücke DNA chemisch-synthetisch herzustellen. Für kurze Stücke von 20 bis 50 Nukleotiden Länge geht das ganz gut und man nennt diese Stücke „Primer„.
Nimmt man nun eine lange einzelsträngige DNA und gibt einen Primer dazu, der an einer Stelle „passt“ (d.h. Basenpaare ausbilden kann und ein kurzes Stück Doppelstrang bildet), dann verlängert die DNA-Polymerase den Primer nach der Vorlage des Einzelstrangs und wir haben am Ende einen langen Doppelstrang. Voraussetzung ist natürlich, dass der Polymerase genügend Bausteine (Nukleotide) zur Verfügung stehen, um sich die richtigen für die Verlängerung des Primers auszusuchen.
Nun ist DNA aber fast immer doppelsträngig (Doppelhelix). Wie macht man daraus Einzelstränge? In der Zelle machen das Helikasen, das sind Enzyme, die den Doppelstrang aufwinden. Im Reagenzglas funktioniert das aber nicht gut. Der Doppelstrang geht aber auch ganz einfach durch Hitze auf. Wenn man die DNA auf 90°C erhitzt, lösen sich die Wasserstoffbrücken, die die gegenüberliegenden Nukleotide verbinden und aus dem Doppelstrang werden zwei Einzelstränge. Gibt man dann Primer dazu und kühlt das Gefäß ab, findet der Primer genau die Stelle an die er passt und bildet dort Wasserstoffbrücken aus. Man kann ausrechnen, wie groß die Wahrscheinlichkeit ist, dass in einer bestimmten DNA der Primer nur eine einzige Stelle findet, an die er genau passt. So weiß man, dass ein Primer mit 20 Nukleotiden Länge statistisch nur eine exakte Bindungsstelle in einem menschlichen Genom mit 3,3 Milliarden Basenpaaren hat.
Bis hier können wir also folgendes: DNA, Nukleotide und Primer in ein Gefäß geben, erhitzen, etwas abkühlen, Polymerase zugeben und warten bis die Polymerase ihren Job gemacht hat.
Will man das wiederholen, taucht das nächste Problem auf: wenn wir das Gefäß wieder erhitzen, wird die DNA wieder einzelsträngig, Primer und Nukleotide stört die Hitze auch nicht – aber die Polymerase ist ein Protein und Proteine mögen normalerweise keine Hitze (wir wissen, dass aus einem gekochten Ei kein Küken schlüpfen kann!).
Aber auch dazu wurde eine Lösung gefunden: In heißen Quellen hatte man Bakterien entdeckt, die sich bei Umgebungstemperaturen von 50 bis 80°C richtig wohlfühlten. Die mussten also eine besondere DNA-Polymerase haben, der Hitze nichts ausmacht. Es dauerte nicht so lange, bis man die Polymerase fand und das Protein in großen Mengen in Bakterien produzieren konnte. Das Bakterium hatte den Namen Thermus aquaticus bekommen und die Polymerase ist seitdem als Taq-Polymerase bekannt. Damit war es möglich die Primerverlängerungsreaktion mehrmals hintereinander durchzuführen, ohne dass die Polymerase den Geist aufgab.

Das Spiel ging aber noch weiter. Wenn man nicht einen, sondern zwei Primer nimmt, von denen einer auf den „unteren“ und einer auf den „oberen“ Strang der DNA passt, läuft die Verlängerung zwischen den beiden Primern hin und her und das DNA Stück dazwischen wird jedes Mal verdoppelt. Nach einer Reaktionsrunde hat man 2 Stücke, nach zwei Runden vier, nach drei Runden acht usw. Nach 30 Runden (Zyklen) gibt es ca. 1 Milliarde Kopien von dem Stück zwischen den beiden Primern (230). In der Abbildung ist das noch einmal verständlich dargestellt. Wer Lust hat, kann sich auch den Film ansehen, in dem noch ein paar weitere Details gezeigt sind.
Aber was kann man nun mit 1 Milliarden DNA Stücken anfangen?
Damit geht es im nächsten Beitrag weiter und dann kommen wir auch zu den Mördern und Vätern, von denen Lukas geschrieben hat.
Hier sind die drei wesentlichen Schritte der PCR nochmals zusammengefasst:

1. Denaturierung
Die Doppelhelix wird durch Erhitzen auf 90°C in Einzelstränge geschmolzen. Ursache ist das Lösen von Wasserstoffbrückenbindungen zwischen den Basenpaaren.
2. Annealing
Unter „Annealing“ versteht man das spezifische Anlagern der Primer an ihre Zielsequenz. Das Anlagern beruht auf der Ausbildung von Wasserstoffbrücken zwischen den komplementären Basenpaaren. Dabei muss für jedes Primerpaar die optimale Annealingtemperatur bestimmt werden. Sie darf nicht zu niedrig gewählt werden, da es sonst zu unspezifischen Anlagerungen kommt, darf aber auch nicht zu hoch gewählt werden, da die Primer sich sonst wieder vom Einzelstrang lösen. Für die Berechnung stehen verschiedene Formeln zur Verfügung, die mehr oder weniger Parameter berücksichtigen. Vereinfacht hängt sie von der Länge der Primer und ihrer Nucleotidsequenz ab und liegt irgendwo zwischen 50 und 70°C.
3. Elongation:
Die „Elongation“ beschreibt den Prozess der Kettenverlängerung. Die DNA-Polymerase synthetisiert – ausgehend vom jeweiligen Primer – mit den freien Nucleotiden den fehlenden Strang. Bei einer Temperatur von 72°C, welches für die Taq-Polymerase die optimale Arbeitstemperatur darstellt, polymerisiert sie etwa 1.000 Nucleotide in einer Minute.